
Enfermedad de Stargardt
Es la distrofia macular más frecuente. La mayoría de casos se heredan de forma autosómica recesiva. La disminución de visión que produce esta enfermedad afecta habitualmente a personas jóvenes. La incidencia de la enfermedad de Stargardt se sitúa alrededor de una persona afectada entre 10.000 personas y suele afectar a adolescentes y adultos jóvenes menores de 20 años. La enfermedad de Stargardt y el fundus flavimaculatus son dos presentaciones clínicas de la misma enfermedad. A nivel histológico se produce un cúmulo de material tipo lipofuscina en las células del epitelio pigmentario de la retina por la mutación del gen ABCA4. Esta mutación se transmite cuando los dos progenitores también poseen la malformación de este gen.
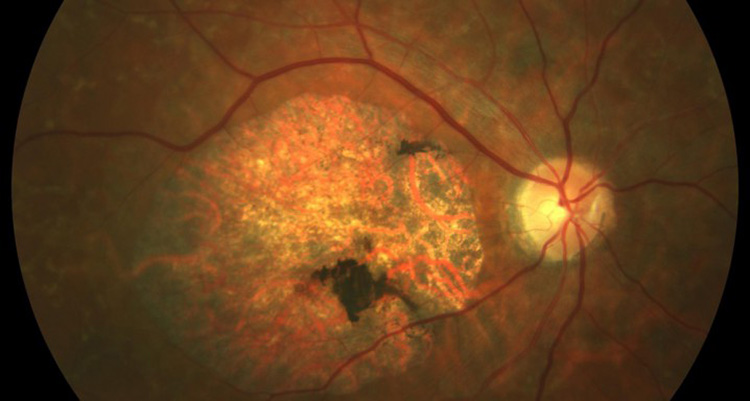
La enfermedad de Stargardt provoca una visión desenfocada y sin nitidez, que dificulta reconocer rostros y formas así como leer tanto de cerca como de lejos, además al final induce a confundir colores de matices próximos (por ejemplo, negro y azul marino)
También causa dificultad para adaptarse a la penumbra. Aunque no provoca ceguera absoluta, las personas que la padecen pueden perder agudeza visual hasta llegar a la ceguera legal.
Los pacientes con esta patología se subdividen en cuatro grupos:
- El primero, caracterizado por el fondo ocular color bronce y silencio coroideo, es el estadio más precoz de la enfermedad. El fondo del ojo es prácticamente normal excepto por el típico silencio coroideo en la angiofluoresceingrafía.
- En el segundo grupo, la maculopatía atrófica con o sin flecos amarillentos, inicialmente la pérdida de EPR (Epitelio Pigmentario de la Retina) puede ser tan mínima que en algunos pacientes sólo se pone de manifiesto al hacer la angiografía.También la aparición de fondo color bronce y silencio coroideo puede no ser evidente en la primera década de la vida. Sin embargo, más tarde aparecen las lesiones amarillentas y el almacenamiento de lipofuscina se hace evidente. El grado y el patrón de la zona de atrofia en el área macular varían y no siempre se correlaciona con el grado de pérdida visual. Es muy común que aparezca un patrón en ojo de buey, que en algunos casos les permite conservar agudezas visuales de unidad hasta la edad de 40 años. Estos pacientes pueden desarrollar en ocasiones cambios periféricos a nivel del EPR y ocasionalmente pueden desarrollar membranas neovasculares subretinianas que producen lesiones disciformes en la mácula. El test de visión de colores normalmente muestra una alteración leve en el eje rojo-verde. Muchos pacientes muestran un alargamiento de la adaptación a la oscuridad.
- El tercer grupo se refiere a pacientes com maculopatía atrófica con signos y síntomas tardíos de retinitis pigmentosa. Estos son similares a los del segundo grupo pero en edades más tardías de la vida aparecen síntomas y signos de retinitis pigmentosa incluyendo la ceguera nocturna y anormalidades del ERG (Electroretinograma) fotópico (en condiciones de luz y que traduce disfunción de conos) y escotópico (en condiciones de oscuridad que traduce disfunción de bastones). Actualmente se considera que estos pacientes padecen realmente una distrofia de conos y bastones producida por mutaciones severas en ABCA4.
- El cuarto grupo de pacientes afectados por la enfermedad presentan flecos amarillentos no asociados a atrofia macular. Es el cuadro clínico descrito como fundus flavimaculatus. Estos pacientes pueden tener lesiones amarillentas centrales y paracentrales asociadas con mínima evidencia angiográfica o fundoscópica de atrofia del EPR entre estas lesiones. Suele haber silencio coroideo.
La agudeza visual puede ser normal si el centro de la fóvea no está afectado por una de estas lesiones, aunque muchos pacientes tienen un gran fleco en la foveola y agudeza visual disminuida. En ausencia de información del resto de la familia y en ojos con no muy clara evidencia de silencio coroideo puede ser muy difícil o imposible de diferenciarlos con una distrofia en patrón simulando fundus flavimaculatus.
Dr. Jordi Monés, M.D., Ph.D.Número de Colegiado COMB: 22.838
Director Médico de la BMF
Doctor en Medicina y Cirugía
Especialista en Oftalmología
Especialista en Retina, Mácula y Vítreo
La información proporcionada en el sitio web no reemplaza sino que complementa la relación entre el profesional de la salud y su paciente o visitante y en caso de duda se debe consultar con su profesional de la salud de referencia.
La BMF desarrolla un programa de ensayos clínicos para la búsqueda de nuevos tratamientos. ¡Contáctanos! Puedes ser el paciente que buscamos.
Quiero tomar parte en los ensayos clínicosContenido destacado de la Enfermedad de Stargardt



La investigación es la única solución de futuro para luchar contra la ceguera
Sólo con tu ayuda lo podremos hacer posible. ¡Ayúdanos a ayudarte!
Colabora