
Malaltia de Stargardt
És la distròfia macular més freqüent. La majoria de casos s’hereten de manera autosòmica recessiva. La disminució de visió que comporta aquesta malaltia afecta habitualment persones joves. La incidència de la malaltia de Stargardt se situa entorn d’ una persona afectada entre 10.000 i sol afectar adolescents i adults joves menors de 20 anys. La malaltia de Stargardt i el fundus flavimaculatus són dos presentacions clíniques de la mateixa malaltia. A nivell histològic, es produeix un cúmul de material tipus lipofuscina a les cèl·lules de l’epiteli pigmentari de la retina per la mutació del gen ABCA4. Aquesta mutació es transmet quan els dos progenitors també posseeixen la malformació d’aquest gen.
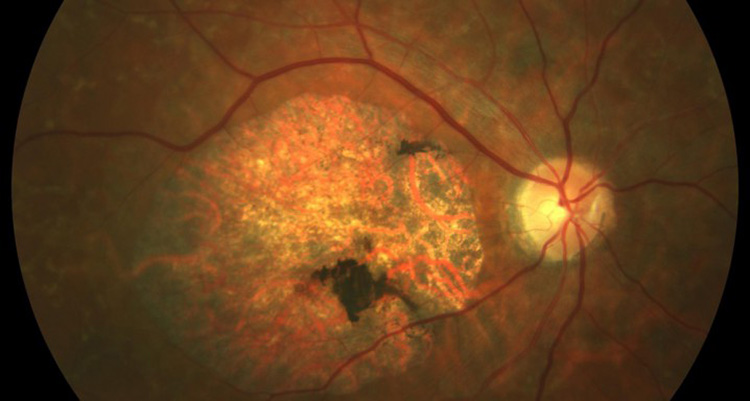
La malaltia de Stargardt provoca una visió desenfocada i sense nitidesa, que dificulta reconèixer rostres i formes, i llegir tant de prop com de lluny i que al final indueix a confondre colors de matisos propers (per exemple, negre i blau marí).
També causa dificultat per adaptar-se a la penombra. Encara que no provoca ceguesa absoluta, les persones que la pateixen poden perdre agudesa visual fins a arribar a la ceguesa legal.
Els pacients amb aquesta patologia se subdivideixen en quatre grups:
- El primer, caracteritzat pel fons ocular color bronze i silenci coroïdal, és l’estadi més precoç de la malaltia. El fons d’ull és pràcticament normal excepte pel típic silenci coroïdal a l’angiofluoresceingrafia.
- En el segon grup, la maculopatia atròfica amb o sense serrells groguencs, inicialment la pèrdua d’ERP (Epiteli Pigmentari de la Retina) pot ser tan mínima que en alguns pacients només es posa de manifest al fer l’angiografia.
- El tercer grup es refereix a pacients amb maculopatia atròfica amb signes i símptomes tardans de retinosi pigmentària. Aquests són similars als del segon grup però en edats més tardanes de la vida apareixen símptomes i signes de retinosi pigmentària incloent la ceguesa nocturna i anormalitats de l’ERG (Electroretinograma) fotòpic (en condicions de llum i que tradueix disfunció de cons) i escotòpic (en condicions d’obscuritat que tradueix disfunció de bastons). Actualment es considera que aquests pacients pateixen realment una distròfia de cons i bastons produïda per mutacions severes en ABCA4.
- El quart grup de pacients afectats per la malaltia presenten serrells groguencs no associats a atròfia macular. És el quadre clínic descrit com fundus flavimaculatus. Aquests pacients poden tenir lesions groguenques centrals i paracentrals associades amb mínima evidència angiogràfica o fundoscòpica d’atròfia de l’ERP entre aquestes lesions. Sol presentar-se silenci coroïdal.
L’agudesa visual pot ser normal si el centre de la fòvea no està afectat per una d’aquestes lesions, tot i que molts pacients tenen un gran serrell a la fovèola i agudesa visual disminuïda. En cas de mancança d’informació de la resta de la família i davant d’ulls amb poca evidència de silenci coroïdal pot ser molt difícil o impossible diferenciar-los amb una distròfia en patró simulant fundus flavimaculatus.
Dr. Jordi Monés, M.D., Ph.D.
Número de Col·legiat COMB: 22.838
Director Mèdic BMF
Doctor en Medicina i Cirurgia
Especialista en Oftalmologia
Especialista en Retina, Màcula i Vitri
La informació proporcionada al lloc web no reemplaça sinó que complementa la relació entre el professional de la salut i el seu pacient o visitant i en cas de dubte s'ha de consultar amb el seu professional de la salut de referència.
La BMF desenvolupa un programa d'assaigs clínics per a la recerca de nous tractaments. Contacta’ns! Pots ser el pacient que busquem.
Vull prendre part en els assaigs clínicsContingut destacat amb la Malaltia d'Stargardt


«El camí per controlar malalties com la DMAE o Stargardt està traçat, però es necessiten molts recursos per fer investigació»

LITE - Ús integrat de Microscòpia de Generació de Segon Harmònic (SHG) per veure fibril•les de col•lagen corni i Oftalmoscòpia Làser d'Escaneig amb Òptica Adaptativa (AOSLO) per veure els fotoreceptors
La recerca és l'única solució de futur per lluitar contra la ceguesa
Només amb el teu ajut ho podrem fer possible. Ajuda'ns a ajudar-te!
Col·labora-hi